Прогнозування з перших принципів стабільних фаз у системі Mg–Rh–B–H
Chem. Met. Alloys 10 (2017)
99-105
https://doi.org/10.30970/cma10.0363
В.І. ІВАЩЕНКО1, І.Ю. ЗАВАЛІЙ2*, П.Е.А. ТУРЧІ3, Н.Р.
МЕДЮХ1, Ю.В. ВЕРБОВИЦЬКИЙ2
1 Інститут проблем матеріалознавства, НАН України, вул. Кржижанівського
3, 03142 Київ, Україна
2 Фізико-механічний інститут, НАН України, вул. Наукова 5, 79060 Львів, Україна
3 Національна лабораторія Лоуренс Лівермор (L-352), P.O. Box 808,
Лівермор, Каліфорнія 94551, США
* Контактна особа. Тел.: +380-32-2296833; e-mail: zavaliy@ipm.lviv.ua
Проведено дослідження з перших
принципів можливих структур системи Mg–Rh–B–H. Початкові кристалічні структури
фаз Mg–Rh–B–H, отримані шляхом заповнення пустот у структурі типу Ti2Ni
атомами бору або / та гідрогену, було згодом оптимізовано. Оптимальні
кристалічні структури визначено на основі аналізу енергій утворення та густини
електронних станів. Стабільними фазами вважалися такі, в яких атоми бору
утворюють структурні одиниці B@Rh4, а атоми гідрогену утворюють H@RhMg3
та H@Rh4. Обговорено вірогідні причини стійкості структур Mg–Rh–B–H.
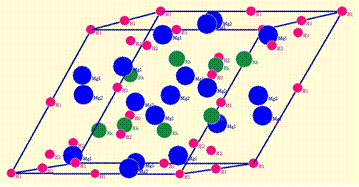
Зменшена ромбоедрична комірка для
Mg16Rh8H02H14H28H32.
Ключові слова
Розрахунки з перших принципів / Кристалічна
структура / Електронний стан / Інтерметаліди / Гідрид