First-principles
prediction of stable phases in the Mg–Rh–B–H system
Chem.
Met. Alloys 10 (2017)
99-105
https://doi.org/10.30970/cma10.0363
V.I. Ivaschenko, I.Yu. Zavaliy,
P.E.A. Turchi, N.R. Medukh, Yu.V. Verbovytskyy
A first-principles investigation of plausible
structures of the Mg–Rh–B–H system was carried out.
The initial crystal structures of the Mg–Rh–B–H
phases obtained by filling the voids in the Ti2Ni structure type by
boron or/and hydrogen atoms, were subsequently optimized. The optimum crystal
structures were determined on the basis of an analysis of formation energies
and densities of electronic states. The stable phases were predicted to be
those in which the boron atoms form B@Rh4 units, and the hydrogen
atoms form H@RhMg3 and H@Rh4 units. Plausible origins of
the stability of the Mg–Rh–B–H structures are
discussed.
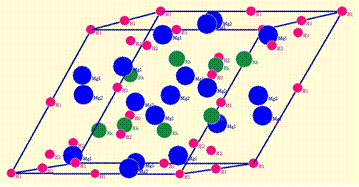
Reduced rhombohedral
cell for Mg16Rh8H02H14H28H32.
Keywords
First-principles
calculations / Crystal structure / Electronic state / Intermetallics / Hydride